EST Exploitation
Jeremy D. Parsons (1997)
Introduction
Expressed Sequence Tags (ESTs)
have grown in utility and importance in recent years as they have become
more numerous,
and better connected to other biological data. ESTs have been mapped onto
portions of the human and other genomes leading to transcript
maps (and on to gene
maps) or just assembled together to form sequence contigs with similarity
to better understood biological sequences. There are many
groups doing the sequencing work and others
add value as the information flows into the public databases. Whilst the
exploitation of ESTs has been improving,
there are still many opportunities to push the public data even further
by increasing its intrinsic quality (e.g. via
assembly), its connectedness (to other databases), and its presentation.
Planned Work
Quality
To improve the intrinsic quality of EST sequence information, I propose
to use a new base-calling program: Phred,
on all the publicly available EST
trace data . Phred was written by Phil Green at Washington University
and improved over the last four years to the point where it has now been
shown to produce fewer errors than the original ABI
basecalls which until recently have been almost universally accepted. Of
greater significance, Phred can produce a base quality file that defines
which basecalls can be trusted and which cannot. Where the original traces
are not available, it should be possible to estimate basecall quality by
counting 'N' calls and by comparing sample sequences to the EMBL mRNA databases
to measure typical error rates and distributions.
EST Overlap-Detection
ESTs, as samples from a larger cDNA, can come in many positions and either
5', or 3' orientations. In addition, each gene may have many alternative
transcripts so it is not a trivial task to assign each EST to its progenitor
gene even with perfect EST sequencing. As this is a difficult problem,
it makes sense to use as much information as possible when trying to determine
the relationship of EST to gene. The main sources of ancillary information
are basecall quality measures or estimates, forward and reverse read linkage,
clone sizing experiments, and library source data to help distinguish polymorphisms
and sequencing errors from gene homologues.
I propose to use a new dynamic programming-based sequence alignment
algorithm that can use much of the ancillary information directly in its
calculations. Based on a no-end-gap global similarity comparison with penalties
moderated by quality etc. it should be possible to obtain sequence alignments
and scores that reflect the true likelihood of any two ESTs being derived
from the same original transcript. This should be better than the local
similarity sequence alignment used in Phil Green's Phrap
assembly program which is seen as the current benchmark. The overlap detection
process should not be confused with normal database similarity searching
where the focus is on sensitivity because selectivity is more important
when identifying and screening putative overlaps.
Dynamic programming is slow and the potential number of sequence overlaps
huge (Order N*N) so the comparisons will be pre-filtered by a partial indexing
scheme similar to that used in Acembly
(and in the creation of UniGene)
and the detailed alignments will be distributed using PVM
so CPU power should not be limiting.
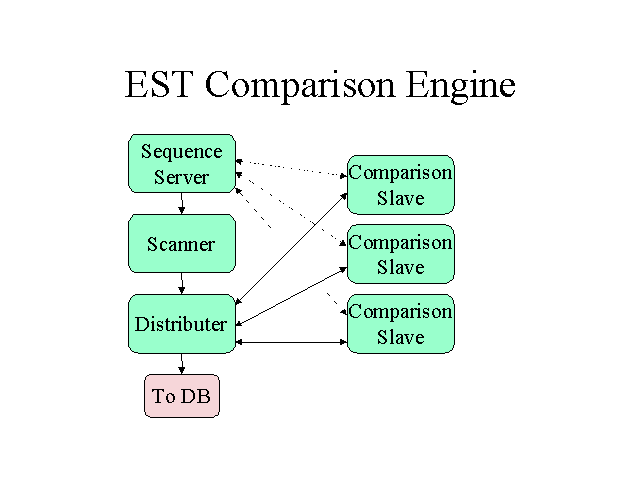
To test the sequence alignment process, the initial focus is just the
detection of EST to EST overlaps but if the tests show that this is producing
high-quality alignments (relative to EMBL mRNA sequences) the overlaps
idea could be extended to other EMBL DNA sequences. Prescreening of ESTs
similar to known mRNAs is more likely to be useful and a simpler means
of data integration
Overlap Database
The ultimate goal of this work is to connect as many different biological
data sources together to as large an extent as possible through ESTs from
maps to functions and whole genome views or specialized views. At the moment
however much of the biolological information and many of the database links
are not present. One step above the raw, submitted data view is a database
of sequence overlaps. If this database is up to date, accurate and easily
accessible, it may provide a new base for contig assembly programs or any
other application where sequence overlap, not homology, is of interest.
One example of such a program is the Glaxo WWW interface to BLASTN to allow
EST assembly in remote sites that have no database searching or assembly
facilities of their own. Most of the problems with the Glaxo approach could
be fixed by a pre-computed database of validated overlaps, some putative
assemblies and some local client interactivity. To accommodate a variety
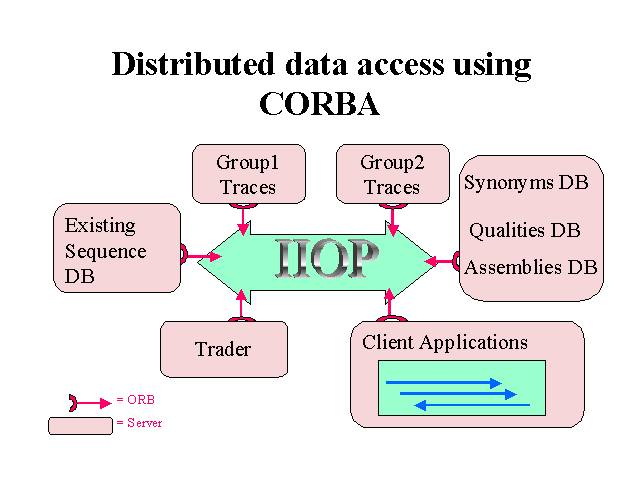
of client interactions with the database, it should be built to allow CORBA
access. Storing all the calculated overlaps allows the database to
be kept up to date without repeated computation effort so as a design advantage,
the database would exist in some form anyway, and any extra burdon for
external access should be minimal.
Of direct interest to researchers may be a classification of ESTs to
reveal those that have no known overlaps on either of their ends. These
reads should directly lead on to novel sequence information if they are
re-sequenced from the publicly available clones or via some form of PCR.
Contig and Consensus Databases
It can be relatively simple to determine that two DNA sequences share enough
similarity to suggest that they could be derived from the same original
mRNA but it is a much more difficult problem to determine whether they
are genuinely cognate. To work towards that goal, various additional constraints
can be applied to the overlap information to determine which of any possible
EST assemblies is judged the most likely. Ultimately, biological experiments
may be necesssary to confirm any contig constructions but suggesting best-guesses
and ranges of options could still be extremely valuable. Any whole-database
assembly can enforce the constraint that each EST may only appear in one
contig. Adding restriction digest, and cDNA sizing information and protein
coding region predictions/constraints can further guide the assembly towards
the correct answer. These guiding data can be combined through the use
of a directed graph and some kind of weighted simulated
annealing algorithm.

Once a set of putative assemblies has been created, they can be given
to the user community for annotation and improvement, or used directly
to generate consensus sequences and then translated to generate an adjunct
to the TREMBL database. This will require new applets or applications.
Links to Washington University's trace server combined with the applets
and the overlap database will provide biologists at remote sites with a
functionality equivalent to having their own bioinformatics team on site.
By allowing users the opportunity to review all the decisions made by the
automatic assembly in a contig viewer, and pairwise alignment editor, their
confidence can be increased or errors more quickly discovered and then
corrected.
If an assembly joins both 5' and 3' reads of a long enough original
clone, it should connect both 3' untranslated regions which are often used
as the basis of STSs, and the upstream sequence which is more likely to
be coding and therefore more likely to have been ascribed a putative function.
Thus, the better the assemblies created, the better the linkage between
functions, genes and map locations. For those contigs that are shorter
than their putative cDNA, it may still be possible to use 3' clustering
to confirm the relationship between the sequence fragments and create gapped
assemblies where some of the target cDNA is missing.
Any assemblies that reveal alternative contig constructions or high
quality sequence mismatches would be ideal starting points for researchers
interested in human polymorphism detection.
EST Data Links
In 1995 Francis Collins predicted (in Nature) that the positional
candidate approach would become the "predominant method for cloning human
diseases". Since it is the desire to understand and ultimately cure human
diseases that is driving most of the current wave of genome science, it
would not be inappropriate to guide the views of the available human genetic
information towards greater support of the Positional Candidate (PC) approach.
This requires genes to be both mapped, to localize them approximately in
the genome, and also sequenced, sufficient to suggest their cellular function
or at least guide experiments towards the latter. It has been estimated
(Schuler et al, Science, 1996) that the majority of the estimated 100,000
human genes have already been sampled and made available in the public
EST databases. and over 16,000 EST gene based STS have been mapped already
so ESTs currently provide the best link between gene function and location.
This link can be improved by creating better EST assemblies that connect
unique untranslated portions of cDNAs to their coding regions which are
more likely to have similarity to genes of known function in the same or
other organisms.
Physical maps can be prepared from end-sequencing, fingerprinting, cross
hybridization, STS amplimer detection, and radiation hybrid mapping. An
immediate goal is a gene map that integrates the sequence map and the physical
maps into a single consistent view. Through the use of CORBA it should
be possible to standardise access to all the component data sources and
thereby ease the data integration task. Putative translations of EST assemblies
can also be linked to the protein databases by amino acid sequence similarity.
EST Data Presentation
Using interactive Java clients to view complex biological data has become
the norm with genome viewers for GDB,
and microbial genomes (JMGD),
physical maps (e.g.
X), and transcript maps (e.g. human
Chr.21). As many of these tools share similar functions, a BioWidgets
consortium has been established to maximise the amount of code-reuse, quality
and availability of the Java clients. I expect most of the required viewers will be written
by others as Java Beans
minimising the actual work needed.
The three basic tools needed are a contig assembly editor, a trace viewer
and a pairwise alignment viewer. A more sophisticated map browsing tool
will follow on to summarise and display all the information combined together
in an integrated gene map. One desirable feature not currently available
anywhere would be a multiple genome gene map. This could provide an ideal
mechanism for browsing areas of syteny between the human and mouse genomes
which both have large EST, mapping and disease genetics information stores
available, but are currently kept and displayed separately.